
Junying Yuan's Team Uncovers Microglial Inflammatory "Brake Mechanism" Offering New Insights for Alzheimer’s and Comorbidities
Source:Xingxing Xie
2026-03-03
On February 2, 2026, the team led by Academician Junying Yuan from the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, published a paper online in the top journal Immunity, systematically clarifying the key role of the INPP5D–RIPK1 signaling axis in regulating microglial inflammatory homeostasis and neurodegenerative diseases.
INPP5D is a major genetic risk factor for late-onset Alzheimer’s disease (LOAD), and RIPK1 is a core molecule mediating neuroinflammation, yet their regulatory mechanisms in microglia remained unclear. The study found that INPP5D directly binds to phosphorylated RIPK1 via its N-terminal SH2 domain, inhibiting RIPK1 kinase activity and serving as a critical "brake" for microglial immune homeostasis.
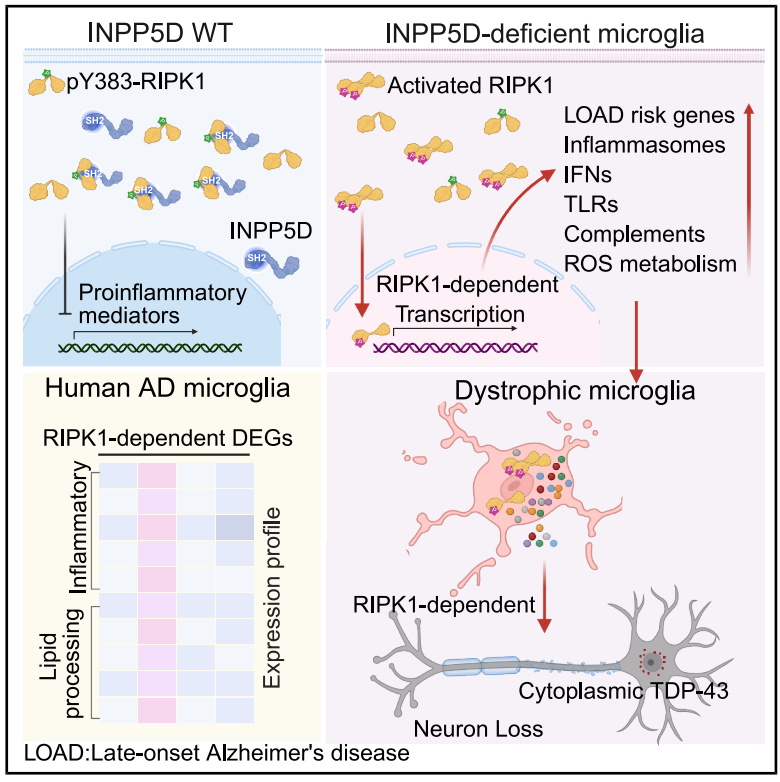
Loss of INPP5D function triggers abnormal RIPK1 activation, leading to amplified inflammatory responses, upregulation of LOAD risk genes, and activation of multiple proinflammatory pathways. It also promotes RIPK1-dependent neuronal TDP-43 pathology in an aging context, causing neuronal loss, motor dysfunction, and progressive neurodegeneration linked to AD-ALS comorbidity.
INPP5D acts as an "intracellular rheostat" for microglial inflammation: its dysregulation amplifies chronic inflammation and drives aging-related neurodegenerative diseases. This discovery deepens the understanding of LOAD genetic risk genes, provides a theoretical basis for RIPK1-targeted interventions, and offers a novel perspective for deciphering AD-ALS comorbidity and developing treatments for intractable neurodegenerative diseases.
Article Link: https://www.cell.com/immunity/fulltext/S10747613(26)00036-1
INPP5D is a major genetic risk factor for late-onset Alzheimer’s disease (LOAD), and RIPK1 is a core molecule mediating neuroinflammation, yet their regulatory mechanisms in microglia remained unclear. The study found that INPP5D directly binds to phosphorylated RIPK1 via its N-terminal SH2 domain, inhibiting RIPK1 kinase activity and serving as a critical "brake" for microglial immune homeostasis.
Loss of INPP5D function triggers abnormal RIPK1 activation, leading to amplified inflammatory responses, upregulation of LOAD risk genes, and activation of multiple proinflammatory pathways. It also promotes RIPK1-dependent neuronal TDP-43 pathology in an aging context, causing neuronal loss, motor dysfunction, and progressive neurodegeneration linked to AD-ALS comorbidity.
INPP5D acts as an "intracellular rheostat" for microglial inflammation: its dysregulation amplifies chronic inflammation and drives aging-related neurodegenerative diseases. This discovery deepens the understanding of LOAD genetic risk genes, provides a theoretical basis for RIPK1-targeted interventions, and offers a novel perspective for deciphering AD-ALS comorbidity and developing treatments for intractable neurodegenerative diseases.
Article Link: https://www.cell.com/immunity/fulltext/S10747613(26)00036-1
