
Lei Shen’s Group Reveals the Mechanism of LKB1 Regulating Insulin Resistance
Source:Lei Shen
2024-07-08
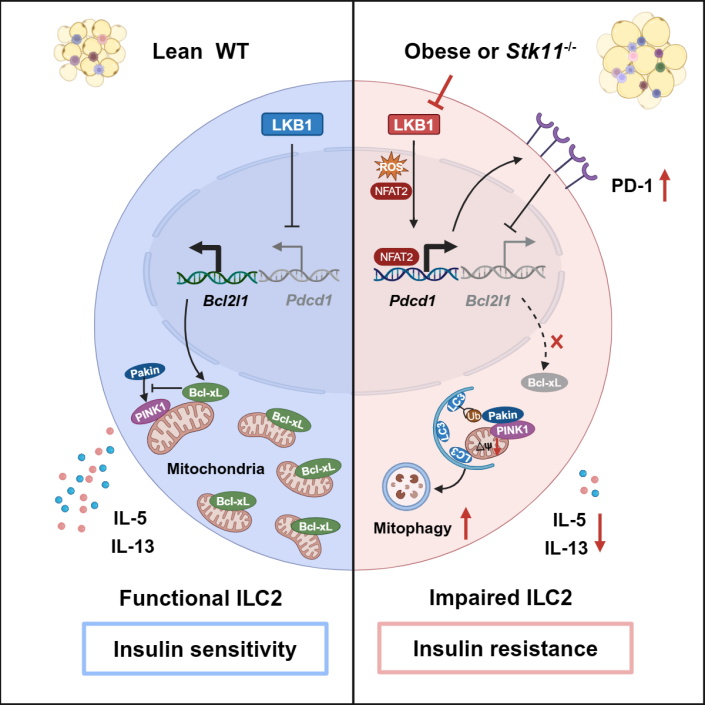
Obesity is associated with numerous complications such as insulin resistance, which often precedes type 2 diabetes (T2D). Although multiple environmental and genetic factors contribute to the development of obesity-induced insulin resistance, low-grade chronic inflammation of adipose tissue is considered a major determinant. Group 2 innate lymphoid cells (ILC2s) play important roles in maintaining adipose tissue homeostasis. They sustain type 2 immunity by recruiting eosinophils and activating alternatively activated macrophages (M2) through production of IL-4, IL-5, and IL-13 cytokines. Additionally, ILC2s control adipose tissue metabolism by promoting beiging through the release of methionine-enkephalin peptides or IL-4/IL-13 that increase UCP1 expression in adipocytes. Targeting ILC2s has become an important strategy for the treatment of obesity-related metabolic diseases. During obesity, the ILC2 compartment is disrupted in both humans and mice, primarily demonstrated by the reduced frequency and decreased IL-5 and IL-13 production in adipose tissue ILC2s. However, the mechanisms underlying such disruption remain largely unknown.
On May 20, 2024, Lei Shen’s research group at Shanghai Institute of Immunology, Shanghai Jiao Tong University School of Medicine published a paper titled “Metabolic regulator LKB1 controls adipose tissue ILC2 PD-1 expression and mitochondrial homeostasis to prevent insulin resistance” in the journal Immunity. This study found that ILC2s employed LKB1 signaling to maintain mitochondrial fitness and effector functions through inhibiting PD-1 expression. Ablation of LKB1 specifically in ILC2s resulted in insulin resistance by suppressing type 2 cytokine production and cell survival.
Through RNAseq analysis, Shen’s group found that adipose tissue ILC2s from high-fat diet (HFD)-induced obese mice exhibited impaired mitochondrial metabolism and decreased expression of liver kinase B1 (LKB1), a key regulator of cell metabolism. To determine whether LKB1 has a functional role in regulating ILC2s, They generated Il5RFP-cre (R5/+) Stk11f/f mice to specifically delete LKB1 in ILC2s and found that LKB1 deficiency led to decreased cell number of VAT ILC2s, and reduced IL-5 and IL-13 production. Furthermore, R5/+ Stk11f/f mice exhibited increased adipose inflammation and exacerbated obesity-induced insulin resistance. Mechanistically, LKB1 deficiency induced aberrant PD-1 expression through activation of ROS-NFAT signaling, which in turn enhanced mitophagy by suppressing Bcl-xL expression.
The study also revealed that PD-1 blockade ameliorates insulin resistance in obese mice. Using ILC2 adoptive transfer experiment and ILC2-specific depletion mouse model (Nmur1icre-tdtDTRf/+), they confirmed that the therapeutic effect of PD-1 blockade on insulin resistance was dependent on ILC2s. Interestingly, They also observed impaired LKB1 signaling and increased PD-1 expression in ILC2s from omentum of obese T2D patients. Moreover, PD-1 blockade restored effector function of ILC2s in omentum of diabetic patients. Overall, these studies reveal the molecular mechanism by which ILC2 function was impaired during obesity and indicate that LKB1-PD-1 axis is a potential therapeutic target for the treatment of obesity associated metabolic disorders.
Professor Lei Shen from Shanghai Institute of Immunology is the corresponding author of this paper. Associate research fellow Jiping Sun and PhD student Youqin Zhang at Shanghai Institute of Immunology are co-first authors of the paper. The research was funded by the National Natural Science Foundation of China, the Ministry of Science and Technology of China, and Science and Technology Commission of Shanghai Municipality.
Links: https://www.sciencedirect.com/science/article/pii/S1074761324002292
On May 20, 2024, Lei Shen’s research group at Shanghai Institute of Immunology, Shanghai Jiao Tong University School of Medicine published a paper titled “Metabolic regulator LKB1 controls adipose tissue ILC2 PD-1 expression and mitochondrial homeostasis to prevent insulin resistance” in the journal Immunity. This study found that ILC2s employed LKB1 signaling to maintain mitochondrial fitness and effector functions through inhibiting PD-1 expression. Ablation of LKB1 specifically in ILC2s resulted in insulin resistance by suppressing type 2 cytokine production and cell survival.
Through RNAseq analysis, Shen’s group found that adipose tissue ILC2s from high-fat diet (HFD)-induced obese mice exhibited impaired mitochondrial metabolism and decreased expression of liver kinase B1 (LKB1), a key regulator of cell metabolism. To determine whether LKB1 has a functional role in regulating ILC2s, They generated Il5RFP-cre (R5/+) Stk11f/f mice to specifically delete LKB1 in ILC2s and found that LKB1 deficiency led to decreased cell number of VAT ILC2s, and reduced IL-5 and IL-13 production. Furthermore, R5/+ Stk11f/f mice exhibited increased adipose inflammation and exacerbated obesity-induced insulin resistance. Mechanistically, LKB1 deficiency induced aberrant PD-1 expression through activation of ROS-NFAT signaling, which in turn enhanced mitophagy by suppressing Bcl-xL expression.
The study also revealed that PD-1 blockade ameliorates insulin resistance in obese mice. Using ILC2 adoptive transfer experiment and ILC2-specific depletion mouse model (Nmur1icre-tdtDTRf/+), they confirmed that the therapeutic effect of PD-1 blockade on insulin resistance was dependent on ILC2s. Interestingly, They also observed impaired LKB1 signaling and increased PD-1 expression in ILC2s from omentum of obese T2D patients. Moreover, PD-1 blockade restored effector function of ILC2s in omentum of diabetic patients. Overall, these studies reveal the molecular mechanism by which ILC2 function was impaired during obesity and indicate that LKB1-PD-1 axis is a potential therapeutic target for the treatment of obesity associated metabolic disorders.
Professor Lei Shen from Shanghai Institute of Immunology is the corresponding author of this paper. Associate research fellow Jiping Sun and PhD student Youqin Zhang at Shanghai Institute of Immunology are co-first authors of the paper. The research was funded by the National Natural Science Foundation of China, the Ministry of Science and Technology of China, and Science and Technology Commission of Shanghai Municipality.
Links: https://www.sciencedirect.com/science/article/pii/S1074761324002292
