
Yi Zhou and Wei Chen’s team reveals new roles of ILC3 gut-kidney migration in renal fibrosis
Source:Yi Zhou
2024-06-19
Renal fibrosis is the histological manifestation of progressive and irreversible processes caused by dysfunctional repair to tissue injury and is a common feature in chronic kidney disease (CKD), ultimately leading to end-stage kidney disease. Persistent inflammatory responses triggered by immune cell infiltration are the initiating factors for renal fibrosis. Therefore, an in-depth exploration of inflammatory responses and the roles of immune cells are critical for the clinical prevention and treatment of CKD and renal fibrosis. Innate lymphoid cells (ILCs) are heterogeneous lymphocytes that lack rearranged antigen receptors and react to insults through cytokine production, initiating critical immune responses and tissue repair. Group 3 ILCs (ILC3s) require the expression of retinoid-related orphan receptor γt (RORγt), produce interleukin (IL)-17A and/or IL-22, and are abundant in mucosal barrier sites like the small intestine. ILC3s function in both physiological and pathological processes, including maintenance of mucosal homeostasis, tissue repair, resistance to bacterial infection, and tumor immune surveillance. However, whether ILC3s are involved in renal fibrosis remains unclear.
On June 11, 2024, Professors Yi Zhou and Wei Chen’s research groups from the First Affiliated Hospital of Sun Yat-sen University, and Professor Joseph V. Bonventre’s research group from Harvard Medical School published a paper titled “Intestinal CXCR6+ ILC3s migrate to the kidney and exacerbate renal fibrosis via IL-23 receptor signaling enhanced by PD-1 expression” in the journal Immunity. This study revealed that following kidney injury, intestinal CXCR6+ ILC3s could migrate to the kidneys and activated fibroblasts through an amplified IL-23R/STAT3/RORγt/IL-17A signaling pathway by expressing PD-1, thereby promoting the development of renal fibrosis.
In this study, researchers first examined ILCs in the kidneys of CKD patients and found a significant increase of ILCs in fibrotic kidneys, primarily accumulated within fibrotic niches, with the density correlating positively with the degree of fibrosis. Further analysis of kidney biopsy samples and peripheral blood from CKD patients revealed that ILC3s are the predominant ILC subgroup in fibrotic kidneys, closely associated with the severity of renal fibrosis and kidney function loss. Similar phenomena were observed in various mouse models of renal fibrosis.
Subsequent studies showed a notable reduction of ILC3s in the small intestine correlating with their accumulation in fibrotic kidneys. Using Kaede transgenic mice for in vivo tracking, researchers identified the small intestine as the primary source of renal ILC3s. Moreover, both intestinal and renal ILC3s highly expressed CXCR6, and its ligand CXCL16 was markedly upregulated in the injured renal tubules. Knockout of CXCR6 or blockage of CXCL16 significantly blocked the intestine-kidney migration of ILC3s.
Researchers further explored the roles and mechanisms of ILC3s in renal fibrosis by using experiments of blocking ILC3 migration, bone marrow transplantation, and adoptive transfer. It was found that the profibrogenic roles of ILC3s depended on PD-1 expression. PD-1 bound to IL-23R and inhibited its endocytosis, thereby maintaining IL-23R on the surface of ILC3s and amplifying downstream intracellular JAK2/ STAT3/RORγt signaling and subsequent IL-17A secretion. Ultimately, increased IL-17A drove extracellular matrix production by renal fibroblasts.
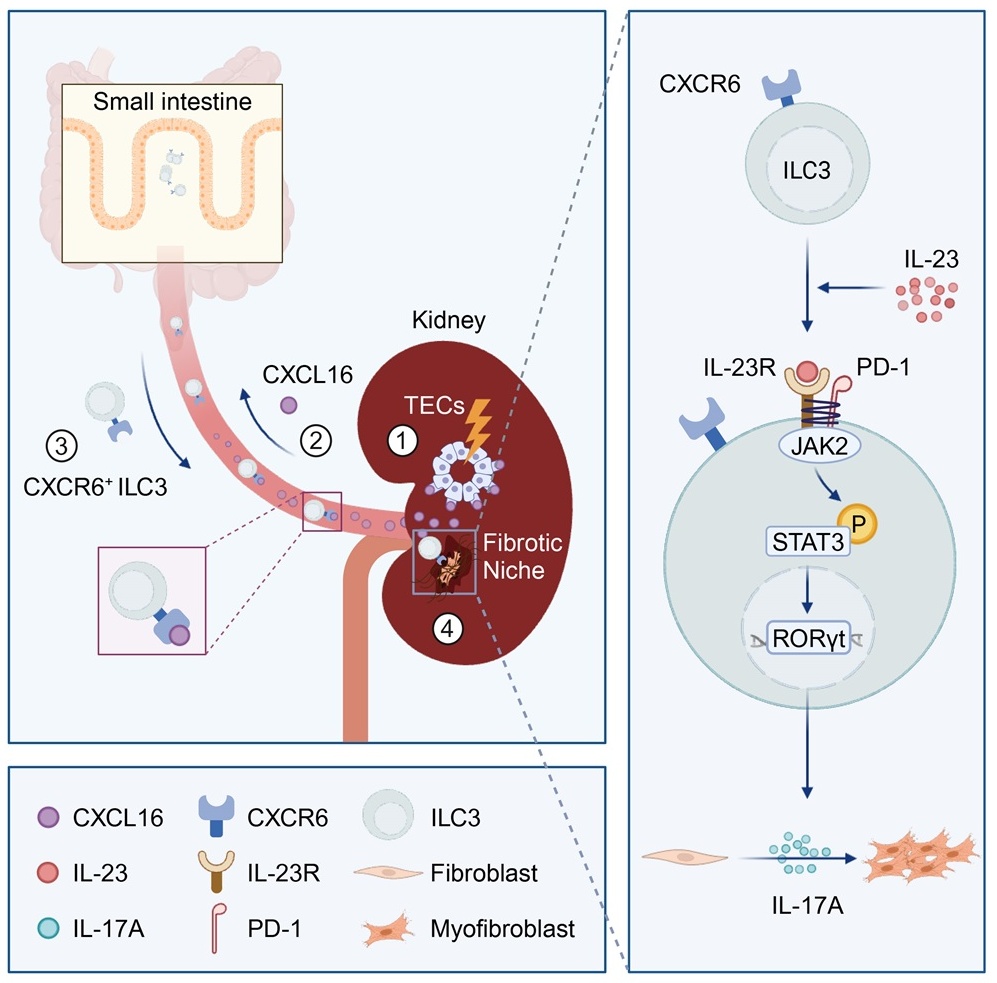
Overall, this study revealed the unique contribution of ILC3s in renal fibrogenesis via the intestine-kidney connection. Targeting ILC3 gut-kidney migration or PD-1 in renal ILC3s has therapeutic potential to prevent CKD.
The First Affiliated Hospital of Sun Yat-sen University is the first affiliated unit for this paper. Professors Yi Zhou, Wei Chen, and Joseph V. Bonventre serve as co-corresponding authors. Postdoctoral researcher Zhou Liang and doctoral students Ziwen Tang and Changjian Zhu are the co-first authors of this paper. The research was funded by the National Natural Science Foundation of China, the NHC Key Laboratory of Clinical Nephrology (Sun Yat-Sen University) and the Guangdong Provincial Key Laboratory of Nephrology, the Guangdong Natural Science Fund,and the Guangdong Special Support Program.
Links: https://doi.org/10.1016/j.immuni.2024.05.004
On June 11, 2024, Professors Yi Zhou and Wei Chen’s research groups from the First Affiliated Hospital of Sun Yat-sen University, and Professor Joseph V. Bonventre’s research group from Harvard Medical School published a paper titled “Intestinal CXCR6+ ILC3s migrate to the kidney and exacerbate renal fibrosis via IL-23 receptor signaling enhanced by PD-1 expression” in the journal Immunity. This study revealed that following kidney injury, intestinal CXCR6+ ILC3s could migrate to the kidneys and activated fibroblasts through an amplified IL-23R/STAT3/RORγt/IL-17A signaling pathway by expressing PD-1, thereby promoting the development of renal fibrosis.
In this study, researchers first examined ILCs in the kidneys of CKD patients and found a significant increase of ILCs in fibrotic kidneys, primarily accumulated within fibrotic niches, with the density correlating positively with the degree of fibrosis. Further analysis of kidney biopsy samples and peripheral blood from CKD patients revealed that ILC3s are the predominant ILC subgroup in fibrotic kidneys, closely associated with the severity of renal fibrosis and kidney function loss. Similar phenomena were observed in various mouse models of renal fibrosis.
Subsequent studies showed a notable reduction of ILC3s in the small intestine correlating with their accumulation in fibrotic kidneys. Using Kaede transgenic mice for in vivo tracking, researchers identified the small intestine as the primary source of renal ILC3s. Moreover, both intestinal and renal ILC3s highly expressed CXCR6, and its ligand CXCL16 was markedly upregulated in the injured renal tubules. Knockout of CXCR6 or blockage of CXCL16 significantly blocked the intestine-kidney migration of ILC3s.
Researchers further explored the roles and mechanisms of ILC3s in renal fibrosis by using experiments of blocking ILC3 migration, bone marrow transplantation, and adoptive transfer. It was found that the profibrogenic roles of ILC3s depended on PD-1 expression. PD-1 bound to IL-23R and inhibited its endocytosis, thereby maintaining IL-23R on the surface of ILC3s and amplifying downstream intracellular JAK2/ STAT3/RORγt signaling and subsequent IL-17A secretion. Ultimately, increased IL-17A drove extracellular matrix production by renal fibroblasts.
Overall, this study revealed the unique contribution of ILC3s in renal fibrogenesis via the intestine-kidney connection. Targeting ILC3 gut-kidney migration or PD-1 in renal ILC3s has therapeutic potential to prevent CKD.
The First Affiliated Hospital of Sun Yat-sen University is the first affiliated unit for this paper. Professors Yi Zhou, Wei Chen, and Joseph V. Bonventre serve as co-corresponding authors. Postdoctoral researcher Zhou Liang and doctoral students Ziwen Tang and Changjian Zhu are the co-first authors of this paper. The research was funded by the National Natural Science Foundation of China, the NHC Key Laboratory of Clinical Nephrology (Sun Yat-Sen University) and the Guangdong Provincial Key Laboratory of Nephrology, the Guangdong Natural Science Fund,and the Guangdong Special Support Program.
Links: https://doi.org/10.1016/j.immuni.2024.05.004
