学术动态
袁钧瑛院士团队《Immunity》解锁小胶质细胞炎症“刹车机制”,为阿尔茨海默症及共病治疗开辟新路径
作者:解醒醒 来源自:中国免疫学会 点击数:16034 发布时间:2026-03-03
2026年2月2日,中国科学院上海有机化学研究所袁钧瑛院士团队在国际顶尖免疫学期刊《Immunity》在线发表高水平研究论文,题为“Repression of RIPK1 kinase by INPP5D inhibits expression of diverse proinflammatory mediators and late-onset Alzheimer’s disease risk factors”。研究首次系统阐明 INPP5D–RIPK1 信号轴在调控小胶质细胞炎症稳态中的核心作用,揭示了其在神经退行性疾病发生发展中的关键机制,为晚发性阿尔茨海默病(LOAD)及相关共病的治疗提供了全新分子靶点和干预思路。
阿尔茨海默病是严重威胁老年人健康的神经退行性疾病,其中晚发性阿尔茨海默病占病例总数的 95% 以上,其发病机制复杂且临床治疗手段有限。现有研究证实,晚发性阿尔茨海默病的遗传风险位点高度富集于小胶质细胞高表达基因中,INPP5D正是晚发性阿尔茨海默病最主要的遗传风险基因之一,但其在小胶质细胞中的具体功能与调控机制长期未被阐明。同时,受体相互作用丝氨酸/苏氨酸蛋白激酶 1(RIPK1)作为介导细胞死亡和神经炎症的关键分子,小胶质细胞中调控其激活稳态的核心机制也始终是领域内的研究空白。
针对上述科学问题,袁钧瑛院士团队展开深入研究,发现INPP5D的N端 SH2结构域可直接结合RIPK1在Tyr383位点的磷酸化修饰,通过抑制 RIPK1 激酶活性,有效限制炎症信号的异常放大,这一分子机制成为小胶质细胞维持中枢免疫稳态的关键“制动装置”,让INPP5D成为调控小胶质细胞炎症强度的“细胞内变阻器”。
研究进一步证实,当小胶质细胞中INPP5D功能缺失时,会引发细胞自主性的RIPK1异常激活,进而触发级联式炎症反应:不仅多种晚发性阿尔茨海默病风险基因被显著上调,促炎细胞因子、补体系统及活性氧相关因子的转录也大幅增强,TLR–MyD88、NLRP3炎症小体等多条促炎信号通路更被系统性激活。团队还在与人类阿尔茨海默病高度相关的小胶质细胞亚型中,鉴定到受RIPK1调控的特异性转录组特征,直接验证了INPP5D–RIPK1信号轴与阿尔茨海默病的病理相关性。
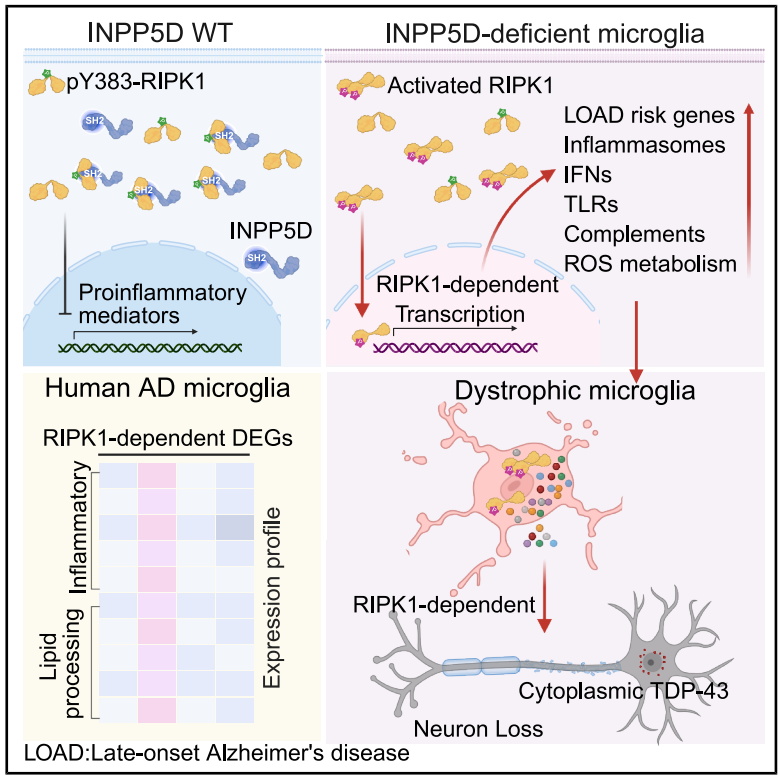
更重要的是,小胶质细胞INPP5D的功能缺陷会通过非细胞自主方式,在衰老背景下促进RIPK1依赖性的神经元TDP-43病理形成,最终导致神经元丢失、小鼠运动功能障碍,并引发与阿尔茨海默病-肌萎缩侧索硬化症(AD–ALS)共病相关的进行性神经退行性改变,这一发现首次为阿尔茨海默病与ALS的共病机制提供了直接的分子证据。
综上,该研究明确了INPP5D在小胶质细胞中通过抑制RIPK1过度激活维持中枢免疫稳态的核心功能;当这一调控体系失衡时,慢性神经炎症被持续放大,最终推动衰老相关神经退行性疾病的发生发展。研究成果不仅深度解析了晚发性阿尔茨海默病遗传风险基因 INPP5D 的功能机制,填补了小胶质细胞炎症稳态调控的领域空白,更首次揭示了INPP5D–RIPK1信号轴作为阿尔茨海默病及相关共病的关键调控节点,为临床靶向RIPK1、重塑小胶质细胞炎症状态开发新型干预策略提供了重要的理论依据和分子靶点,为这类难治性神经退行性疾病的治疗开辟了全新方向。
本研究由中国科学院上海有机化学研究所生物与化学交叉研究中心袁钧瑛院士担任通讯作者,博士后解醒醒为论文第一作者。
论文链接:https://www.cell.com/immunity/fulltext/S10747613(26)00036-1
阿尔茨海默病是严重威胁老年人健康的神经退行性疾病,其中晚发性阿尔茨海默病占病例总数的 95% 以上,其发病机制复杂且临床治疗手段有限。现有研究证实,晚发性阿尔茨海默病的遗传风险位点高度富集于小胶质细胞高表达基因中,INPP5D正是晚发性阿尔茨海默病最主要的遗传风险基因之一,但其在小胶质细胞中的具体功能与调控机制长期未被阐明。同时,受体相互作用丝氨酸/苏氨酸蛋白激酶 1(RIPK1)作为介导细胞死亡和神经炎症的关键分子,小胶质细胞中调控其激活稳态的核心机制也始终是领域内的研究空白。
针对上述科学问题,袁钧瑛院士团队展开深入研究,发现INPP5D的N端 SH2结构域可直接结合RIPK1在Tyr383位点的磷酸化修饰,通过抑制 RIPK1 激酶活性,有效限制炎症信号的异常放大,这一分子机制成为小胶质细胞维持中枢免疫稳态的关键“制动装置”,让INPP5D成为调控小胶质细胞炎症强度的“细胞内变阻器”。
研究进一步证实,当小胶质细胞中INPP5D功能缺失时,会引发细胞自主性的RIPK1异常激活,进而触发级联式炎症反应:不仅多种晚发性阿尔茨海默病风险基因被显著上调,促炎细胞因子、补体系统及活性氧相关因子的转录也大幅增强,TLR–MyD88、NLRP3炎症小体等多条促炎信号通路更被系统性激活。团队还在与人类阿尔茨海默病高度相关的小胶质细胞亚型中,鉴定到受RIPK1调控的特异性转录组特征,直接验证了INPP5D–RIPK1信号轴与阿尔茨海默病的病理相关性。
更重要的是,小胶质细胞INPP5D的功能缺陷会通过非细胞自主方式,在衰老背景下促进RIPK1依赖性的神经元TDP-43病理形成,最终导致神经元丢失、小鼠运动功能障碍,并引发与阿尔茨海默病-肌萎缩侧索硬化症(AD–ALS)共病相关的进行性神经退行性改变,这一发现首次为阿尔茨海默病与ALS的共病机制提供了直接的分子证据。
综上,该研究明确了INPP5D在小胶质细胞中通过抑制RIPK1过度激活维持中枢免疫稳态的核心功能;当这一调控体系失衡时,慢性神经炎症被持续放大,最终推动衰老相关神经退行性疾病的发生发展。研究成果不仅深度解析了晚发性阿尔茨海默病遗传风险基因 INPP5D 的功能机制,填补了小胶质细胞炎症稳态调控的领域空白,更首次揭示了INPP5D–RIPK1信号轴作为阿尔茨海默病及相关共病的关键调控节点,为临床靶向RIPK1、重塑小胶质细胞炎症状态开发新型干预策略提供了重要的理论依据和分子靶点,为这类难治性神经退行性疾病的治疗开辟了全新方向。
本研究由中国科学院上海有机化学研究所生物与化学交叉研究中心袁钧瑛院士担任通讯作者,博士后解醒醒为论文第一作者。
论文链接:https://www.cell.com/immunity/fulltext/S10747613(26)00036-1